Research
Speciation in native slugs

Figure 1: Sister species P. andersoni and P. foliolatum have partially overlapping ranges and differ in ecologically important traits, like microhabitat, foot size, and dentition. A) Map of the Pacific Northwest showing glacial extent during the Last Glacial Maximum (blue), the range of P. andersoni (orange), and the range of P. foliolatum (purple). B) P. andersoni and C) P. foliolatum feeding on mushrooms. D) Comparisons of dentition between P. andersoni and P. foliolatum (Pilsbry & Vanatta 1898).
My lab studies species limits and speciation in native terrestrial gastropods from North America. Taildropper slugs (genus Prophysaon) are endemic to the temperate rainforests of the Pacific Northwest (Figure 1A). There are nine described species, and the group appears to have a complex history with the potential for geology, climate, and ecology to have driven diversification. Our research has supported a likely history of divergence in isolated refugia during glaciation, followed by expansion and gene flow between lineages upon secondary contact in several species (Smith & Carstens, 2020; Smith et al., 2024). Furthermore, we have found evidence of undescribed diversity in this group (Smith et al., 2026).
We are also studying species limits in manteslugs (Genus Philomycus) from the southeastern US. Preliminary results suggest that taxonomic revisions will be needed in this group, as morphology-based identifications often conflict with results from genetic data.

Figure 2: The radula of a specimen of Prophysaon foliolatum. The radula was dissected out and then imaged on a Scanning Electron Microscope at Indiana University.
Machine learning in population genetics
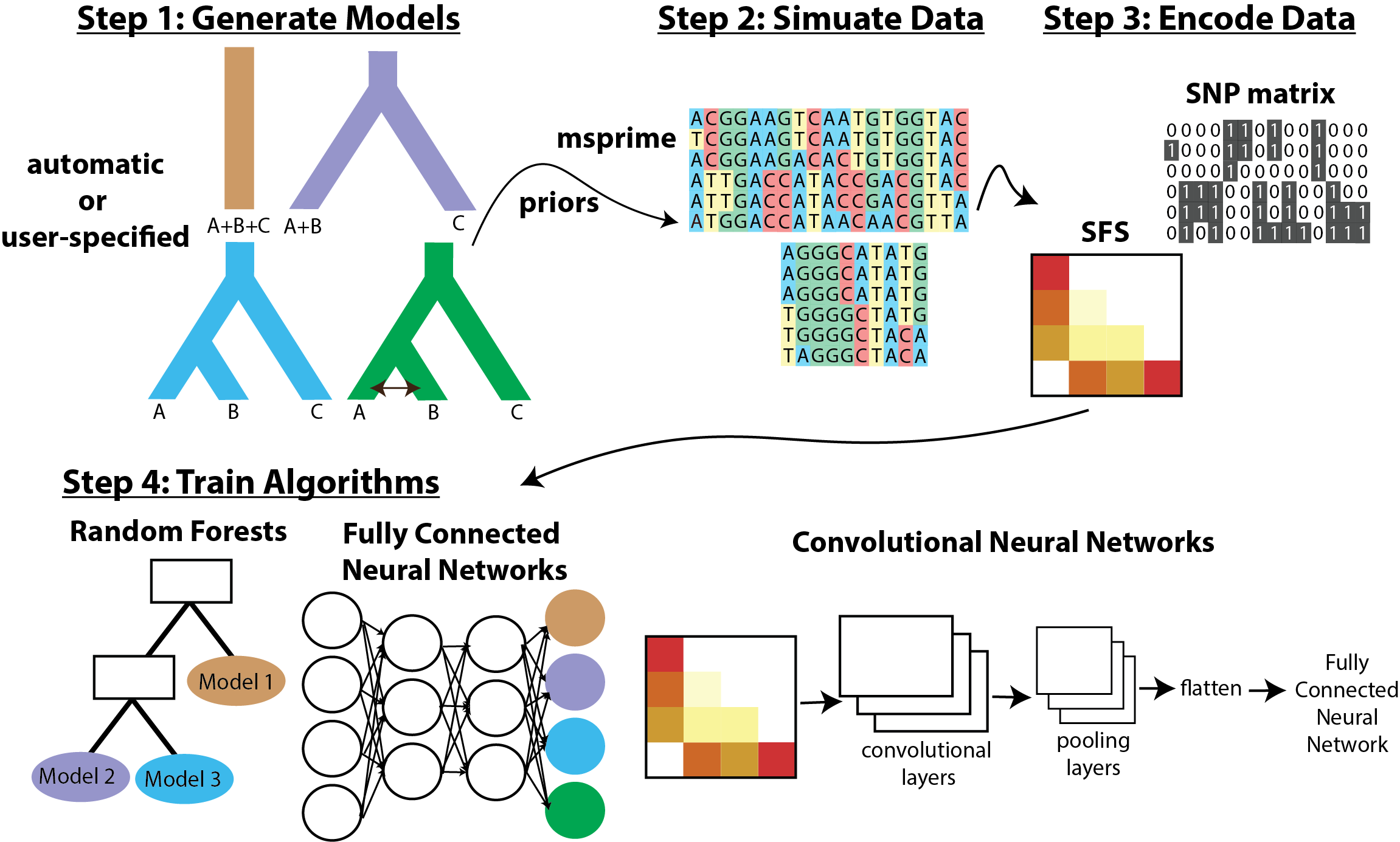
Machine learning approaches are increasingly being applied to answer interesting questions in population genetics and phylogenetics. The lab has developed a python package popai, which infers the evolutionary histories of populations from genomic data using several machine learning approaches. We are also interested in when model violations mislead popular approaches for inferring population histories, detecting selection, and more. For example, my recent work found that selection can mislead inferences of introgression (Smith & Hahn, 2024). We test popular methods using simulated data, use machine learning to identify problematic model violations, and use machine learning approaches (e.g., domain adaptation) to perform more accurate inference in the presence of complex and difficult-to-model biological processes (e.g., background selection or ghost introgression) (e.g., Cobb & Smith, 2025).
New methods for studying gene duplicates
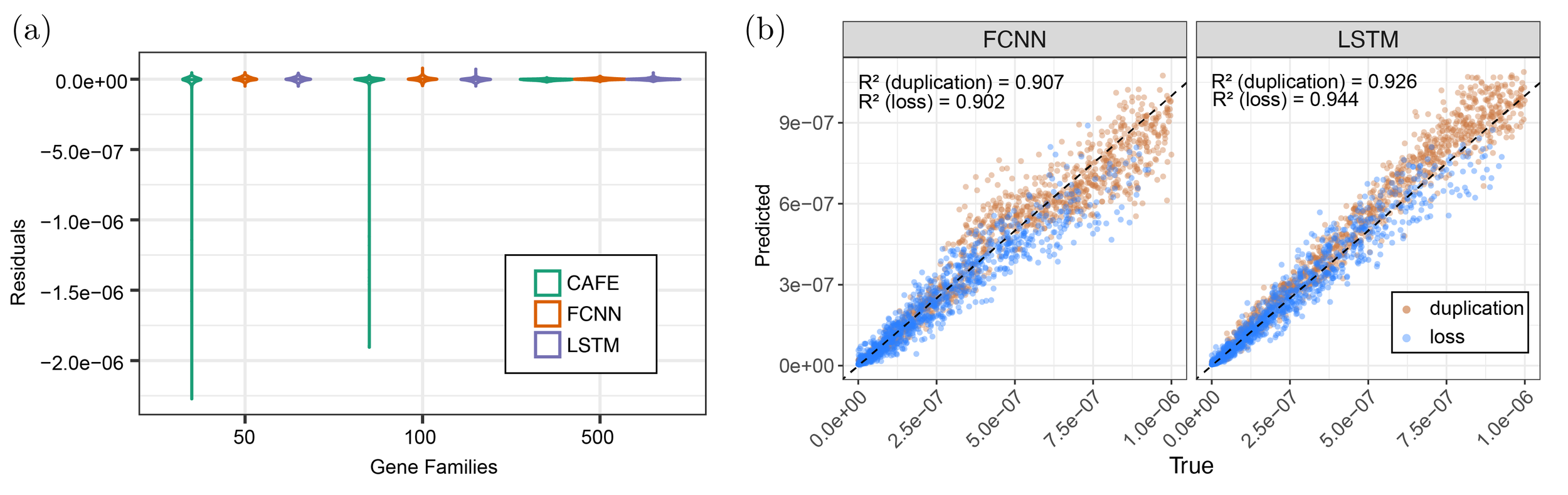
We trained two neural networks to estimate overall rates of gene family evolution or separate duplication and loss rates: a fully connected neural network taking summary statistics and gene family counts as input (FCNN) and an ensemble model with a long-short term network (LSTM) taking lineage-through-time plots as input and an FCNN taking gene family counts as input. (a) Our neural networks outperform CAFE when sample sizes are small and match CAFE in accuracy when sample sizes are larger. (b) Our neural networks can accurately estimate independent duplication and loss rates, a function not available in the newest version of CAFE.
Investigating the invasion histories of invasive terrestrial gastropods
Invasive terrestrial gastropods pose threats to agriculture, human health, and biodiversity, and there are ~70 introduced terrestrial gastropods in the contiguous US. Despite their prevalence, invasive gastropods remain critically understudied, with even basic information including which species are present and the number and sources of introductions remaining unknown. We are both gathering basic, critical info on these invasions and using them as models for studying the evolutionary and ecological causes and consequences of invasions. We are using genomic, phenotypic, and ecological data to study the invasion histories of Arion slugs, Deroceras slugs, and a snail that poses major threats to peanut crops (Bulimulus bonariensis). In Arion and Deroceras our preliminary results support multiple, independent invasions. In some species, species distribution models suggest that habitat filtering is an important determinant of invasive range, while in others, new habitats appear to have been colonized following their introduction to North America.
My lab is interested both in using gene duplicates to improve phylogenetic inference and in understanding the evolutionary dynamics, causes, and consequences of gene duplication and loss. Previously, I investigated the potential benefits and risks of using paralogs (genes related through duplication events) for phylogenetic inference (Yan et al. 2021; Smith and Hahn 2021; Smith and Hahn 2022; Smith et al. 2022). This work highlighted the robustness of phylogenetic inference to the heterogeneity introduced by the inclusion of paralogs and suggests steps towards including more data in phylogenetic analyses. We are also developing a new method, dusti, that infers trees from large gene families directly from alignments—most existing methods use gene trees! More recently, we uncovered a case in which gene duplicates actually improve inference by ameliorating the impacts of long-branch attraction (Smith & Hahn, 2026). We are also developing new machine learning approaches to infer rates of gene duplication and loss, identify lineages and gene families with elevated rates, and investigate relationships between rates of gene duplication and loss, speciation, and phenotypic change.